MetaTiME for Cell State Annotator and Cell Signature Mapper
installation for colab
[4]:
##Calling matplotlib first help avoid error in colab when plotting
import matplotlib
import matplotlib.pyplot as plt
!wget -q https://www.dropbox.com/s/gnmgfxa85basvwq/logo.png
img = matplotlib.image.imread('./logo.png', 0)
logo = plt.imshow( img ) # for test the colab matplotlib. a logo shall be plotted:)
_=plt.axis('off')
## install
!pip install scanpy
!pip install metatime
!pip install anndata
!pip install adjustText
!pip install leidenalg
!pip install harmonypy
imports
[1]:
import sys
import os
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
plt.rcParams['figure.dpi'] = 200
import seaborn as sns
import scanpy as sc
import importlib as imp
import metatime
import matplotlib
import matplotlib.pyplot as plt
"""
## For local testing in dev
METATIME_DIR = '../../metatime/'
if METATIME_DIR not in sys.path:
sys.path.append(METATIME_DIR)
file = '../notebooks_dev/BCC_GSE123813_aPD1_with_metainfo.h5ad'
##
"""
from metatime import config
from metatime import loaddata
from metatime import mecmapper
from metatime import mecs
from metatime import annotator
from metatime import plmapper
from metatime import dmec
import matplotlib
Get sample data
[ ]:
#!wget -O testdata.h5ad https://www.dropbox.com/s/0sj6gtppsfr2ouo/NSCLC_GSE117570_res.pp.h5ad?dl=0
!wget -O testdata.h5ad https://www.dropbox.com/s/f791mv3tnhs9fii/BCC_GSE123813_aPD1_with_metainfo.h5ad
Load scRNA data
[ ]:
file = 'testdata.h5ad'
Or provide path to local file
[3]:
# file = '../notebooks_dev/BCC_GSE123813_aPD1_with_metainfo.h5ad' # Yost et al. 2019 Nat Med.
Load data
[3]:
adata = loaddata.load(file = file)
Loaded ../notebooks_dev/BCC_GSE123813_aPD1_with_metainfo.h5ad
Processing and Harmonize
If your data contains count matrix, we provide a wrapped function for pre-processing the data.
Otherwise, if the data is already depth-normalized, log-transformed, and cells are filtered, we can skip this step.
[21]:
# adata = loaddata.adatapp( adata )
The sample data has multiple patients , and we can use batch correction on patients.
[4]:
batchcols = ['patient']
adata = loaddata.batchharmonize( adata, batchcols = batchcols, random_state = 0 )
[Log ] hamonize with batch ['patient']
2023-03-14 11:18:26,769 - harmonypy - INFO - Iteration 1 of 10
2023-03-14 11:19:26,441 - harmonypy - INFO - Iteration 2 of 10
2023-03-14 11:20:26,466 - harmonypy - INFO - Iteration 3 of 10
2023-03-14 11:21:32,953 - harmonypy - INFO - Iteration 4 of 10
2023-03-14 11:22:26,570 - harmonypy - INFO - Iteration 5 of 10
2023-03-14 11:23:18,462 - harmonypy - INFO - Iteration 6 of 10
2023-03-14 11:24:01,069 - harmonypy - INFO - Iteration 7 of 10
2023-03-14 11:24:29,524 - harmonypy - INFO - Iteration 8 of 10
2023-03-14 11:25:14,176 - harmonypy - INFO - Iteration 9 of 10
2023-03-14 11:26:07,662 - harmonypy - INFO - Iteration 10 of 10
2023-03-14 11:26:58,323 - harmonypy - INFO - Stopped before convergence
[Log ] Recomputing umap
It is recommended that malignant cells are identified first and removed for best practice in cell state annotation.
In the BCC data, the cluster of malignant cells are identified with inferCNV. We can use the pre-saved column ‘isTME’ to keep Tumor Microenvironment cells
[5]:
adata = adata[adata.obs['isTME']]
We can over-cluster the cells which is useful for fine-grained cell state annotation.
[6]:
adata = annotator.overcluster( adata ) # this generates a 'overcluster' columns in adata.obs
/homes6/yiz/Tools/pip/pippy3/lib/python3.7/site-packages/scanpy/tools/_leiden.py:160: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
categories=natsorted(map(str, np.unique(groups))),
Load MetaTiME trained model for TME
Next, let’s load the pre-computed MetaTiME MetaComponents (MeCs), and their functional annotation.
[7]:
# Load the pre-trained MeCs
mecmodel = mecs.MetatimeMecs.load_mec_precomputed()
# Load functional annotation for MetaTiME-TME
mectable = mecs.load_mecname( mecDIR = config.SCMECDIR, mode ='table' )
mecnamedict = mecs.getmecnamedict_ct(mectable)
Project on MeC space
We can project each cell onto this functional MeC space.
[8]:
pdata = mecmapper.projectMecAnn(adata, mecmodel.mec_score)
/liulab/yiz/proj/MetaTiME/metatime/mecmapper.py:151: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour.
adataproj = anndata.AnnData( projected, obs = adata.obs )
Per-cell score projected on MeC space
Convert to table to look at per-cell projection scores
[9]:
projmat, mecscores = annotator.pdataToTable(pdata, mectable, gcol = 'overcluster')
mecscores
[9]:
| 0: Pan_Interferon-response | 1: Pan_Proliferation-S | 2: Pan_Heat-stress | 3: DC_pDC | 4: B_Plasma | 5: Pan_Proliferation-G2-M | 6: DC_cDC2-MHCII | 7: T_Treg-T-co-signaling | 8: T_NK-cytotoxic | 9: M_Monocyte-CD16 | ... | 77: B_Plasma-JCHAIN | 78: T_CD4T-IL7R | MeC_79 | MeC_80 | 81: Stroma_Fibroblast-VEGF | MeC_82 | 83: Pan_Myeloid-fibroblast | MeC_84 | MeC_85 | overcluster | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| bcc.su001.pre.tcell_AAACCTGCAGGGATTG | -0.819890 | -0.688164 | -1.293394 | -0.623175 | -0.619621 | -0.014213 | -0.421470 | -0.575049 | 0.806688 | -0.228148 | ... | -0.503919 | 1.365941 | 0.532829 | 1.303657 | -0.015501 | -0.200811 | 2.227211 | -2.101272 | 0.964769 | 103 |
| bcc.su001.pre.tcell_AAACGGGCATAGACTC | 0.497730 | -0.781702 | -1.236870 | -0.225077 | -0.407063 | -0.196667 | -0.708806 | 0.148677 | 1.226127 | 0.229111 | ... | -0.964028 | -0.869057 | -0.948729 | 0.932158 | -1.617307 | -0.813145 | -0.541694 | -1.985215 | 0.821645 | 71 |
| bcc.su001.pre.tcell_AAACGGGTCATACGGT | -0.853597 | 0.389129 | 2.834898 | -0.235956 | 0.469343 | 0.946867 | -0.673814 | 0.078563 | 1.469857 | 0.388126 | ... | -0.713808 | 1.094122 | 1.178537 | -2.930747 | -2.534621 | 1.399297 | -0.640663 | 2.175453 | -1.674753 | 9 |
| bcc.su001.pre.tcell_AAAGATGAGACAGGCT | 1.643644 | -0.217423 | -0.643116 | 0.088754 | -0.523546 | -1.255705 | -0.849190 | 0.658166 | -0.770305 | 1.756781 | ... | -1.132153 | 1.547834 | -1.547482 | 0.496351 | -1.177201 | -0.764956 | 0.551764 | 0.479404 | -0.165305 | 8 |
| bcc.su001.pre.tcell_AAAGATGTCTGAGTGT | -0.904096 | -0.855004 | -1.025405 | -0.417323 | -0.265433 | -0.517993 | -0.371883 | -0.238967 | -1.225588 | 0.211766 | ... | 0.743283 | 2.142204 | 0.523307 | 0.401327 | -0.243886 | -0.139409 | 1.541129 | -0.646087 | -0.965725 | 40 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| bcc.su012.post.tcell_TTTGCGCGTCTTTCAT | -0.655006 | -0.323290 | 1.357172 | -0.252120 | -0.477362 | -0.562883 | -0.469015 | -0.594926 | -0.978787 | 0.131918 | ... | 0.224142 | 1.476642 | 1.258007 | -0.838055 | 0.543334 | 0.264970 | -0.528499 | 1.782975 | -2.250962 | 19 |
| bcc.su012.post.tcell_TTTGGTTCAGCCAGAA | -1.002944 | -0.177266 | 1.441417 | -0.175024 | -0.643992 | -1.178595 | -1.009185 | 0.391318 | 0.190629 | 1.112531 | ... | 0.111604 | 0.727014 | 0.063260 | 0.221161 | 1.290879 | 0.011502 | -0.428551 | 1.685099 | -0.775345 | 15 |
| bcc.su012.post.tcell_TTTGTCAAGCACCGTC | -0.705487 | -0.384236 | 1.495082 | -0.411420 | -0.401349 | -0.957136 | -1.487738 | 0.004184 | -0.690182 | 0.843381 | ... | -0.196389 | -0.427286 | -0.727919 | -0.666853 | 1.105166 | 3.032268 | -0.136370 | 1.011591 | -0.914623 | 15 |
| bcc.su012.post.tcell_TTTGTCAAGTGAACGC | 0.205688 | -0.372909 | 1.382011 | 0.256174 | -0.391342 | 0.236742 | -1.236226 | 1.282568 | 0.039292 | 0.352591 | ... | -1.496709 | -0.348647 | -0.667981 | 0.685011 | -1.942186 | -0.779483 | -0.759375 | 0.198692 | 0.043316 | 12 |
| bcc.su012.post.tcell_TTTGTCAGTCCCTTGT | -0.059757 | 0.314007 | 1.000176 | -0.132242 | 0.371538 | -0.351378 | -0.189296 | 3.665382 | -0.018753 | 0.335091 | ... | -0.520862 | -0.083217 | 2.131293 | -0.706136 | 0.002483 | -0.122560 | -0.354572 | 1.812793 | -0.942595 | 3 |
38836 rows × 87 columns
Cell state annotator and save the results to scRNA objects
Based on per-cell projection scores, compute the per-cluster cell state scores, and find out the top 1st enriched cell state.
Save the cluster-wise cell state annotation.
Or, it can also be saved to the scanpy object “pdata”, containing only the scores, which is smaller to save.
[10]:
projmat, gpred, gpreddict = annotator.annotator( projmat, mecnamedict, gcol = 'overcluster')
adata = annotator.saveToAdata( adata, projmat )
pdata = annotator.saveToPdata( pdata, adata, projmat )
/liulab/yiz/proj/MetaTiME/metatime/annotator.py:190: UserWarning: Columns in projmat already overlap columns in adata.obs. Overwriting adata.obs.
warnings.warn('Columns in projmat already overlap columns in adata.obs. Overwriting adata.obs. '
New columns added to adata and pdata: - MetaTiME_overcluster
[11]:
## Simplify the cell states as on UMAP plot by updating:
## adata, pdata, mecnamedict
adata.obs['MetaTiME'] = adata.obs['MetaTiME_overcluster'].str.split(': ').str.get(1)
pdata.obs['MetaTiME'] = pdata.obs['MetaTiME_overcluster'].str.split(': ').str.get(1)
mecnamedict_simp = mecnamedict.copy()
_=[mecnamedict_simp.update({t: mecnamedict[t].split(': ')[1]}) for t in mecnamedict.keys()]
New columns added to adata and pdata: - MetaTiME
A simplified naming dictory: - mecnamedict_simp
Plot per-cluster, Top1 enriched cell states
[12]:
fig,ax = plmapper.plot_annotation_on_data( adata, COL = 'MetaTiME', fontsize=7 )
/homes6/yiz/Tools/pip/pippy3/lib/python3.7/site-packages/anndata/_core/anndata.py:1235: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c

Check the list of MeCs
[13]:
mecnamedict_simp
[13]:
{'MeC_0': 'Pan_Interferon-response',
'MeC_1': 'Pan_Proliferation-S',
'MeC_2': 'Pan_Heat-stress',
'MeC_3': 'DC_pDC',
'MeC_4': 'B_Plasma',
'MeC_5': 'Pan_Proliferation-G2-M',
'MeC_6': 'DC_cDC2-MHCII',
'MeC_7': 'T_Treg-T-co-signaling',
'MeC_8': 'T_NK-cytotoxic',
'MeC_9': 'M_Monocyte-CD16',
'MeC_11': 'T_NK-T-cytotoxic',
'MeC_12': 'T_CD8T-GZMK-CCL5',
'MeC_13': 'Myeloid_Mast',
'MeC_14': 'Pan_Proliferation',
'MeC_15': 'Pan_Ribosome',
'MeC_16': 'T_CD4T-MAIT-Th17',
'MeC_17': 'M_Monocyte-CD14',
'MeC_18': 'B_B',
'MeC_19': 'M_Macrophage-C1Q',
'MeC_20': 'T_Tfh-CXCL13',
'MeC_21': 'T_CD4T-cytoskeleton',
'MeC_22': 'Stroma_Fibroblast-collagen',
'MeC_23': 'Stroma_Endothelial',
'MeC_24': 'Pan_Metallothionein',
'MeC_25': 'T_Cytotoxic-GZMB',
'MeC_26': 'M_Macrophage-SPP1',
'MeC_27': 'Stroma_Fibro-endo-EMT',
'MeC_28': 'Pan_Beta-catenin',
'MeC_29': 'M_Monocyte-CD14-S100A12',
'MeC_30': 'B_B-Ig-kappa',
'MeC_31': 'Pan_Protein-processing',
'MeC_32': 'T_CD8T-CD3',
'MeC_33': 'Pan_Cycling-MYC',
'MeC_34': 'T_NK-T-IFNG-CCL4',
'MeC_35': 'T_T-Immune-synapse',
'MeC_36': 'M_Macrophage-SPP1-C1Q',
'MeC_37': 'M_Macrophage-lipid-PPARG',
'MeC_38': 'Stroma_Endothelial-CCL21',
'MeC_39': 'Myeloid_Platelet',
'MeC_40': 'T_CD8Tex-CXCL13',
'MeC_41': 'Stroma_Myofibroblast',
'MeC_42': 'M_Macrophage-IL1-NFKB',
'MeC_43': 'T_T-naive',
'MeC_45': 'M_Macrophage-IL1-JUN',
'MeC_46': 'Myeloid_Erythrocytes',
'MeC_47': 'Pan_Glycolysis',
'MeC_48': 'T_NK-T-CCL5',
'MeC_49': 'M_Macrophage_RNASE1-C1Q',
'MeC_50': 'B_B-PAX5',
'MeC_51': 'DC_DC-Mature',
'MeC_52': 'M_Monocyte-LYZ',
'MeC_53': 'Pan_MHCI',
'MeC_54': 'Pan_Ribosome-RPS18',
'MeC_55': 'DC_cDC1',
'MeC_56': 'T_T-CREM',
'MeC_57': 'Pan_AP1-JUN',
'MeC_58': 'Pan_MHCII',
'MeC_59': 'B_B-Ig-lambda',
'MeC_61': 'Pan_AP1-FOS',
'MeC_63': 'Pan_MHCII-HLA-DRA',
'MeC_64': 'Pan_AP1-IER2',
'MeC_65': 'T_T-co-signaling',
'MeC_66': 'Pan_CCL2',
'MeC_67': 'Pan_Heat-stress-HSPA1A',
'MeC_68': 'Pan_NR4A-signaling',
'MeC_69': 'T_CD4T-CXCR4',
'MeC_74': 'M_Monocyte-CD14-proliferation',
'MeC_75': 'T_NK-T-Cytotoxic-GZMB',
'MeC_76': 'Pan_CXCL1-CXCL2',
'MeC_77': 'B_Plasma-JCHAIN',
'MeC_78': 'T_CD4T-IL7R',
'MeC_81': 'Stroma_Fibroblast-VEGF',
'MeC_83': 'Pan_Myeloid-fibroblast'}
Plot signature continuum for a single MeC
[14]:
fig=plmapper.plot_umap_mec( pdata, 'MeC_3' ,mecnamedict, figfile =None)
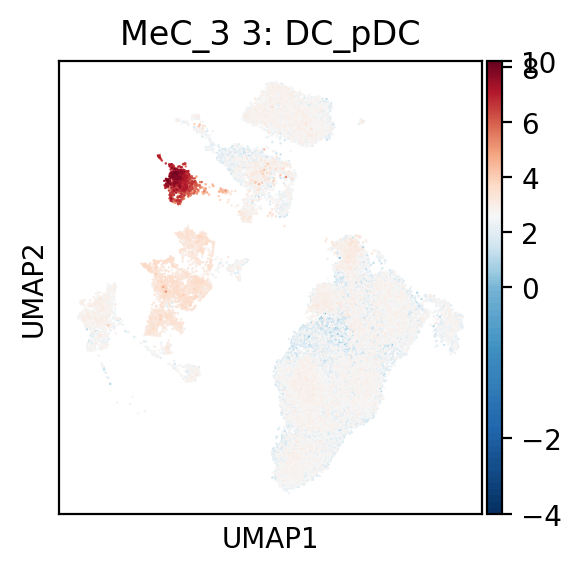
Plot signature continuum for all functional MeCs
[15]:
fig=plmapper.plot_umap_proj_pdata( pdata, mecnamedict , figfile =None)

Comparing conditions
The first way is to count cells newly annotated by MetaTiME. A count table can be returned in all ways grouping the cells. The conditions here are dataset - specific.
[16]:
condcol = 'treatment'
clustercol = 'MetaTiME'
samplecol = 'patient'
cellcts = dmec.count_cell_condition( adata.obs, countbycols = [condcol, clustercol, samplecol] )
#cellcts.to_csv('./cell_counts.txt',sep='\t')
cellcts
[16]:
| patient | su001 | su002 | su003 | su004 | su005 | su006 | su007 | su008 | su009 | su010 | su012 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| treatment | MetaTiME | |||||||||||
| post | B_B | 3447.0 | 0.0 | 8.0 | 19.0 | 55.0 | 18.0 | 2.0 | 127.0 | 2.0 | 0.0 | 0.0 |
| B_B-Ig-kappa | 12.0 | 3.0 | 0.0 | 3.0 | 8.0 | 537.0 | 0.0 | 27.0 | 0.0 | 0.0 | 2.0 | |
| B_B-Ig-lambda | 7.0 | 1.0 | 1.0 | 2.0 | 3.0 | 76.0 | 2.0 | 32.0 | 0.0 | 0.0 | 0.0 | |
| B_B-PAX5 | 273.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| B_Plasma | 15.0 | 4.0 | 0.0 | 5.0 | 16.0 | 164.0 | 2.0 | 23.0 | 8.0 | 0.0 | 2.0 | |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| pre | T_T-CREM | 72.0 | 4.0 | 7.0 | 33.0 | 64.0 | 83.0 | 126.0 | 12.0 | 324.0 | 7.0 | 100.0 |
| T_T-Immune-synapse | 47.0 | 0.0 | 6.0 | 22.0 | 28.0 | 35.0 | 82.0 | 4.0 | 146.0 | 6.0 | 130.0 | |
| T_T-naive | 1.0 | 0.0 | 2.0 | 13.0 | 13.0 | 2.0 | 22.0 | 1.0 | 113.0 | 0.0 | 13.0 | |
| T_Tfh-CXCL13 | 25.0 | 2.0 | 9.0 | 16.0 | 74.0 | 335.0 | 94.0 | 87.0 | 408.0 | 0.0 | 465.0 | |
| T_Treg-T-co-signaling | 200.0 | 6.0 | 8.0 | 52.0 | 79.0 | 300.0 | 166.0 | 90.0 | 338.0 | 3.0 | 353.0 |
76 rows × 11 columns
Writing the cell counts to table
[22]:
cellcts.to_csv('./cell_counts.txt',sep='\t')
MetaTiME also provides functions to test differential signatures cluster-wise.
[17]:
condcol = 'treatment' # the column in adata.obs to mark the condition
cond1 = ['pre'] # Condition 1 in adata.obs[ condcol ],
cond2 = ['post'] # Condition 2 in adata.obs[ condcol ],
clustercol = 'MetaTiME' # MetaTiME annotated cluster labels
# get the cell barcodes for two conditions
allcellscond1= adata[adata.obs[ condcol ].isin(cond1), ].obs.index
allcellscond2= adata[adata.obs[ condcol ].isin(cond2), ].obs.index
[18]:
print(f'Compare cells based on {condcol} with conditions {cond1} and {cond2}')
print(f'Cells in Condition {cond1} : {len(allcellscond1)}')
print(f'Cells in Condition {cond2} : {len(allcellscond2)}')
Compare cells based on treatment with conditions ['pre'] and ['post']
Cells in Condition ['pre'] : 13710
Cells in Condition ['post'] : 25126
Get A table listing all signatures enriching in each cluster, as candidate for testing differential signature.
[19]:
mec_enriched = dmec.enrich_mec(
adata=pdata, labelcol =clustercol, mecnamedict=mecnamedict_simp)
Testing all candidate signatures in each cell cluster.
[20]:
diffmec, diffmecsig, diffmec_full = dmec.dmec(adata, pdata, allcellscond1, allcellscond2, clustercol, mec_enriched, mecnamedict_simp,
test_clusters='all', test_method = 'ttest' )
Plotting the differential signature dot plot.
X-axis: difference of mean signature scores between conditions. Y-axis: -log(p-value) from the test statistic chosen. The significant cluster-wise differential signature is marked as “EnrichedMeC@ClusterName”; when the enriched MeC is the same as cluster name, the signature is marked “ClusterName”. Size of the dots is proportionally to the mean signature score.
[27]:
sns.set_theme(style='white')
fig_topdiff = dmec.plot_topdiff(diffmec, fontsize=6)
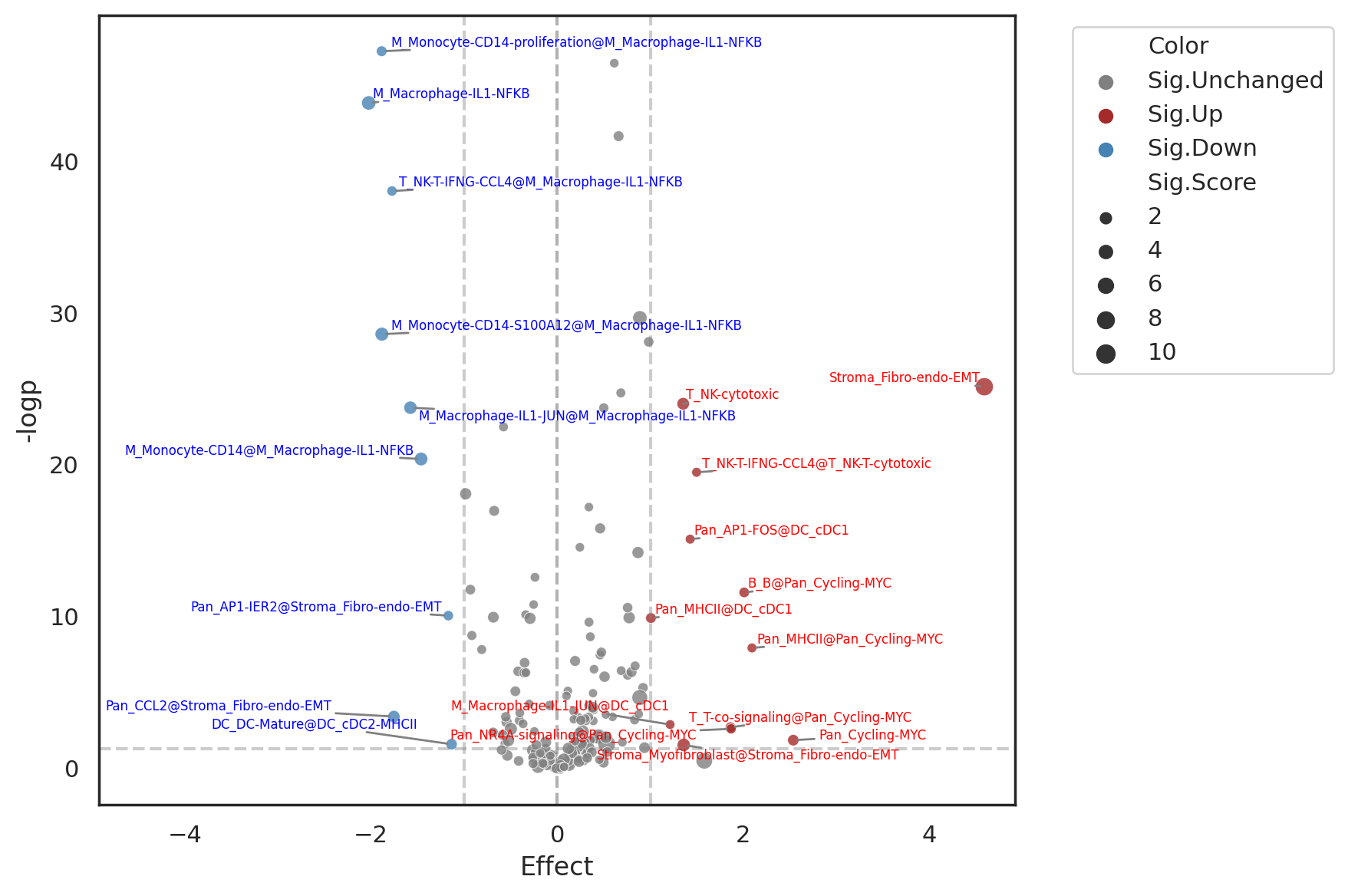
Save all the significant differential signatures@Cluster
[24]:
diff, diff_sig = dmec.topdiff_df(diffmec)
[25]:
diff_sig[:3]
[25]:
| -logp | effect | effect1 | meanb | meana | scorecol | cluster1 | mec | |
|---|---|---|---|---|---|---|---|---|
| M_Monocyte-CD14-proliferation@M_Macrophage-IL1-NFKB | 47.236925 | -1.885173 | -1.885173 | 0.486959 | 2.372133 | MeC_74 | M_Macrophage-IL1-NFKB | M_Monocyte-CD14-proliferation |
| M_Macrophage-IL1-NFKB | 43.831255 | -2.024648 | -2.024648 | 4.083235 | 6.107882 | MeC_42 | M_Macrophage-IL1-NFKB | M_Macrophage-IL1-NFKB |
| T_NK-T-IFNG-CCL4@M_Macrophage-IL1-NFKB | 38.030503 | -1.774028 | -1.774028 | 0.264443 | 2.038471 | MeC_34 | M_Macrophage-IL1-NFKB | T_NK-T-IFNG-CCL4 |
Saving this table
[36]:
diff_sig.to_csv('diff_sig_allcluster.txt',sep='\t')